
Now Reading: ASTROREPOMICS: A Curated Transcriptomic Database For Reproductive Biology In Space
-
01
ASTROREPOMICS: A Curated Transcriptomic Database For Reproductive Biology In Space

ASTROREPOMICS: A Curated Transcriptomic Database For Reproductive Biology In Space
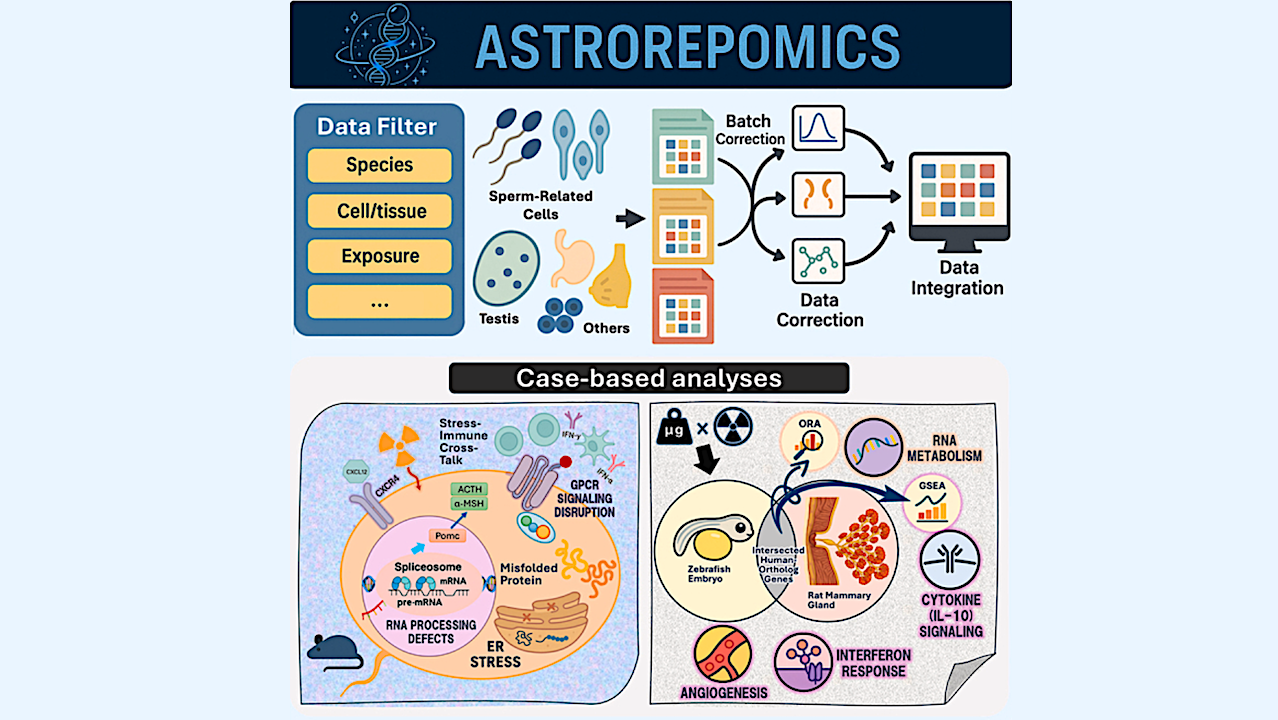
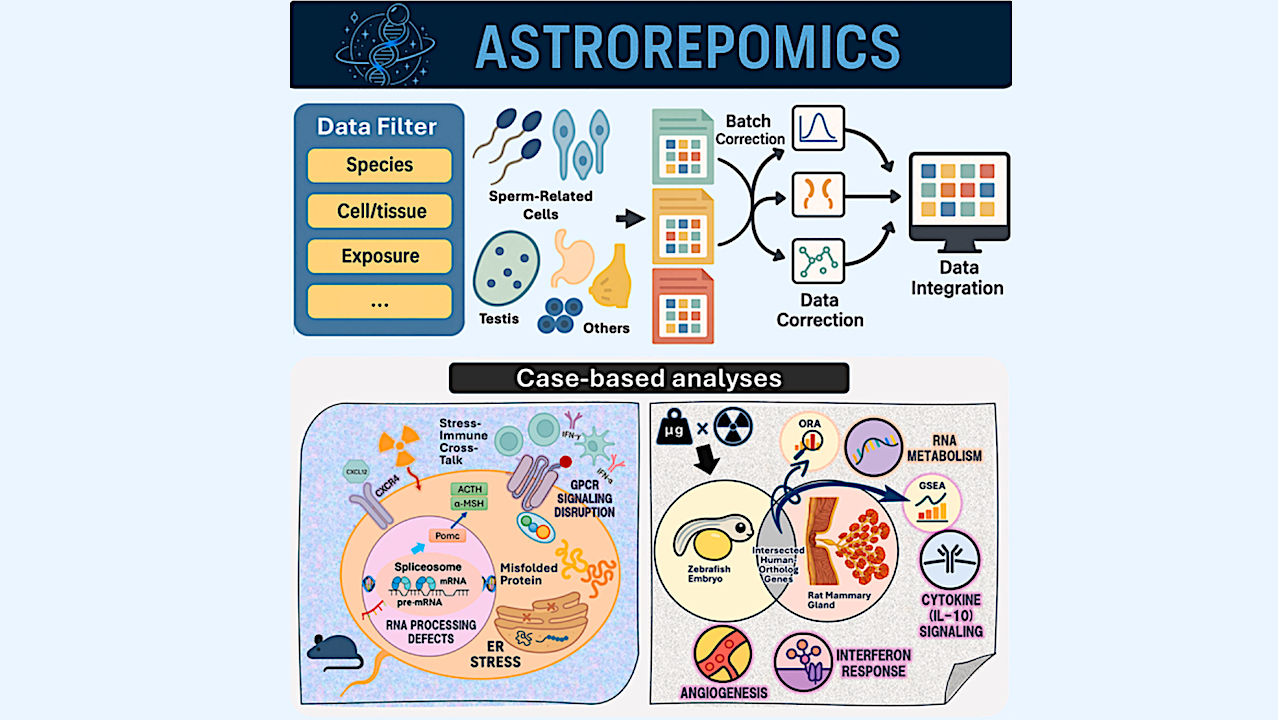
Graphical Abstract — Cell
Spaceflight imposes substantial physiological stress on reproductive systems, yet relevant transcriptomic data remain fragmented across repositories. To address this need, we developed ASTROREPOMICS, a web-based platform that integrates 17 rigorously normalized and batch-corrected transcriptomic datasets spanning multiple species and reproductive tissues. The platform supports reproducible cross-study and cross-species analyses through standardized metadata and an intuitive user interface.
We highlight its utility through two example analyses: (1) irradiated mouse sperm exhibited suppression of RNA splicing and protein-processing pathways alongside activation of interferon- and GPCR-associated programs; and (2) a multi-species intersected-DEG assessment between irradiated rat mammary tissue and microgravity-exposed zebrafish embryos uncovered conserved signatures involving RNA metabolism, cytokine signaling, and angiogenesis.
By consolidating dispersed datasets and offering tailored analytical capabilities, ASTROREPOMICS provides a centralized resource for hypothesis generation and strengthens the research infrastructure needed to advance reproductive health studies in space, supporting long-term efforts to safeguard fertility during deep-space exploration.
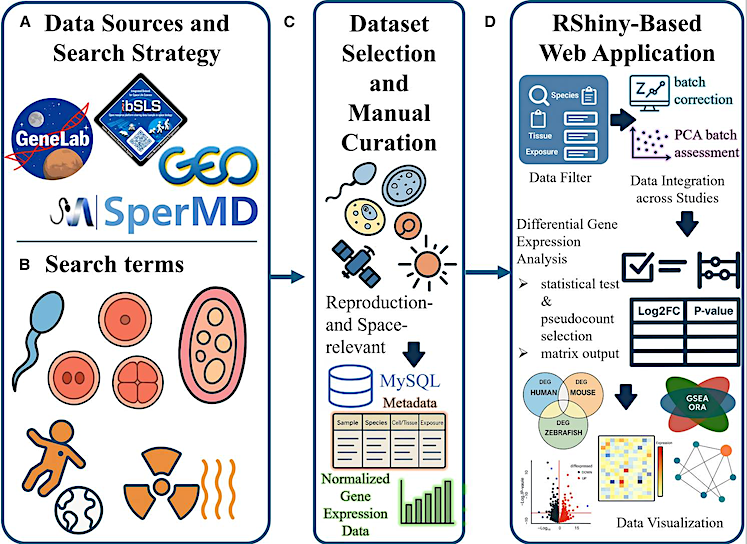
The ASTROREPOMICS workflow — Cell
(A) Transcriptomic datasets were collected from multiple public and institutional repositories, including NASA’s GeneLab, the Gene Expression Omnibus (GEO), the Integrated Biobank for Space Life Science (ibSLS), and the Spermatogenesis-Related Multi-Omics Database (SperMD).
(B) Candidate datasets were retrieved using reproduction-related keywords (e.g., sperm, oocyte, zygote, embryo, and gonadal tissue) and spaceflight-relevant conditions (e.g., microgravity and radiation).
(C) Studies were included only if they were both reproduction- and space-relevant, with metadata completeness confirmed through manual review. Curated datasets were organized within a MySQL-based relational database containing normalized gene expression values and structured sample metadata, including species, tissue or cell type, and exposure condition.
(D) The interface enables user-level access for data filtering, batch correction, and cross-study integration using methods such as per-dataset Z score normalization or ComBat, with principal-component analysis (PCA) provided for assessing and visualizing batch-effect removal. Users can then perform differential gene expression analysis with customizable pseudocounts and statistical tests, followed by downstream visualization. Outputs include cross-species DEG comparison plots, volcano plots, heatmaps, pathway enrichment plots, and pathway-to-gene network diagrams derived from gene set enrichment analysis (GSEA) and over-representation analysis (ORA).
Astrobiology, Genomics, bioinformatics,










Related Posts


Stay Informed With the Latest & Most Important News
Previous Post
Next Post





Advertisement
-
 01Two Black Holes Observed Circling Each Other for the First Time
01Two Black Holes Observed Circling Each Other for the First Time -
 02From Polymerization-Enabled Folding and Assembly to Chemical Evolution: Key Processes for Emergence of Functional Polymers in the Origin of Life
02From Polymerization-Enabled Folding and Assembly to Chemical Evolution: Key Processes for Emergence of Functional Polymers in the Origin of Life -
 03Astronomy 101: From the Sun and Moon to Wormholes and Warp Drive, Key Theories, Discoveries, and Facts about the Universe (The Adams 101 Series)
03Astronomy 101: From the Sun and Moon to Wormholes and Warp Drive, Key Theories, Discoveries, and Facts about the Universe (The Adams 101 Series) -
 04True Anomaly hires former York Space executive as chief operating officer
04True Anomaly hires former York Space executive as chief operating officer -
 05Φsat-2 begins science phase for AI Earth images
05Φsat-2 begins science phase for AI Earth images -
 06Hurricane forecasters are losing 3 key satellites ahead of peak storm season − a meteorologist explains why it matters
06Hurricane forecasters are losing 3 key satellites ahead of peak storm season − a meteorologist explains why it matters -
 07Binary star systems are complex astronomical objects − a new AI approach could pin down their properties quickly
07Binary star systems are complex astronomical objects − a new AI approach could pin down their properties quickly